Real cancer drivers walkthrough
Source:vignettes/v02_real_cancer_drivers.Rmd
v02_real_cancer_drivers.RmdReal cancer drivers walkthrough
This vignette uses the bundled
dataset_real_cancer_drivers_4 dataset to illustrate a real
biological analysis: how do four canonical cancer driver catalogs
overlap?
The four sources are:
- Vogelstein — the 138-gene catalog from Vogelstein et al. (Science 2013), often cited as the “core” oncogene set.
- COSMIC_CGC — the COSMIC Cancer Gene Census (Sondka et al. 2018), a curated list of genes causally implicated in cancer.
- OncoKB — the MSK precision-oncology knowledge base annotation level ≥ “Oncogenic” (Chakravarty et al. 2017).
- IntOGen — pan-cancer driver mutations from the IntOGen pipeline (Martínez-Jiménez et al. 2020).
library(vennDiagramLab)
ds <- load_sample("dataset_real_cancer_drivers_4")
ds@set_names
#> [1] "Vogelstein" "COSMIC_CGC" "OncoKB" "IntOGen"Set sizes
sapply(ds@items, length)
#> Vogelstein COSMIC_CGC OncoKB IntOGen
#> 138 581 1231 633The lists are very different in size — Vogelstein is the smallest curated set; OncoKB is the most permissive at this annotation tier.
Universe
The dataset was built from a 20,000-gene background
(universe_size):
ds@universe_size
#> [1] 20000This is the population N used in the hypergeometric
over-representation tests (see
vignette("v05_statistics_deep_dive")).
Set sizes (inclusive) and intersection layout
result@set_sizes
#> Vogelstein COSMIC_CGC OncoKB IntOGen
#> 138 581 1231 633A summary at a glance
broom::glance() returns a one-row tibble with the
headline numbers:
broom::glance(result)
#> # A tibble: 1 × 8
#> n_sets n_regions n_items universe_size model is_approximate n_significant
#> <int> <int> <int> <int> <chr> <lgl> <int>
#> 1 4 15 20000 20000 venn-4-set FALSE 6
#> # ℹ 1 more variable: n_highly_significant <int>Render the venn diagram
The default render uses the dataset’s set names as labels. To shorten them for the diagram, pass a per-letter override:
svg <- render_venn_svg(
result,
set_names = c(A = "Vogelstein", B = "COSMIC", C = "OncoKB", D = "IntOGen"),
title = "Cancer driver overlap (4 sources)"
)
nchar(svg)
#> [1] 6475(See vignette("v08_custom_styling_and_export") for color
overrides and post-render SVG manipulation.)
UpSet view
For 4+ sets, an UpSet plot is often easier to read than the Venn diagram — each intersection size is a bar, sorted by cardinality.
upset_plot <- render_upset(result, sort_by = "size")
upset_plot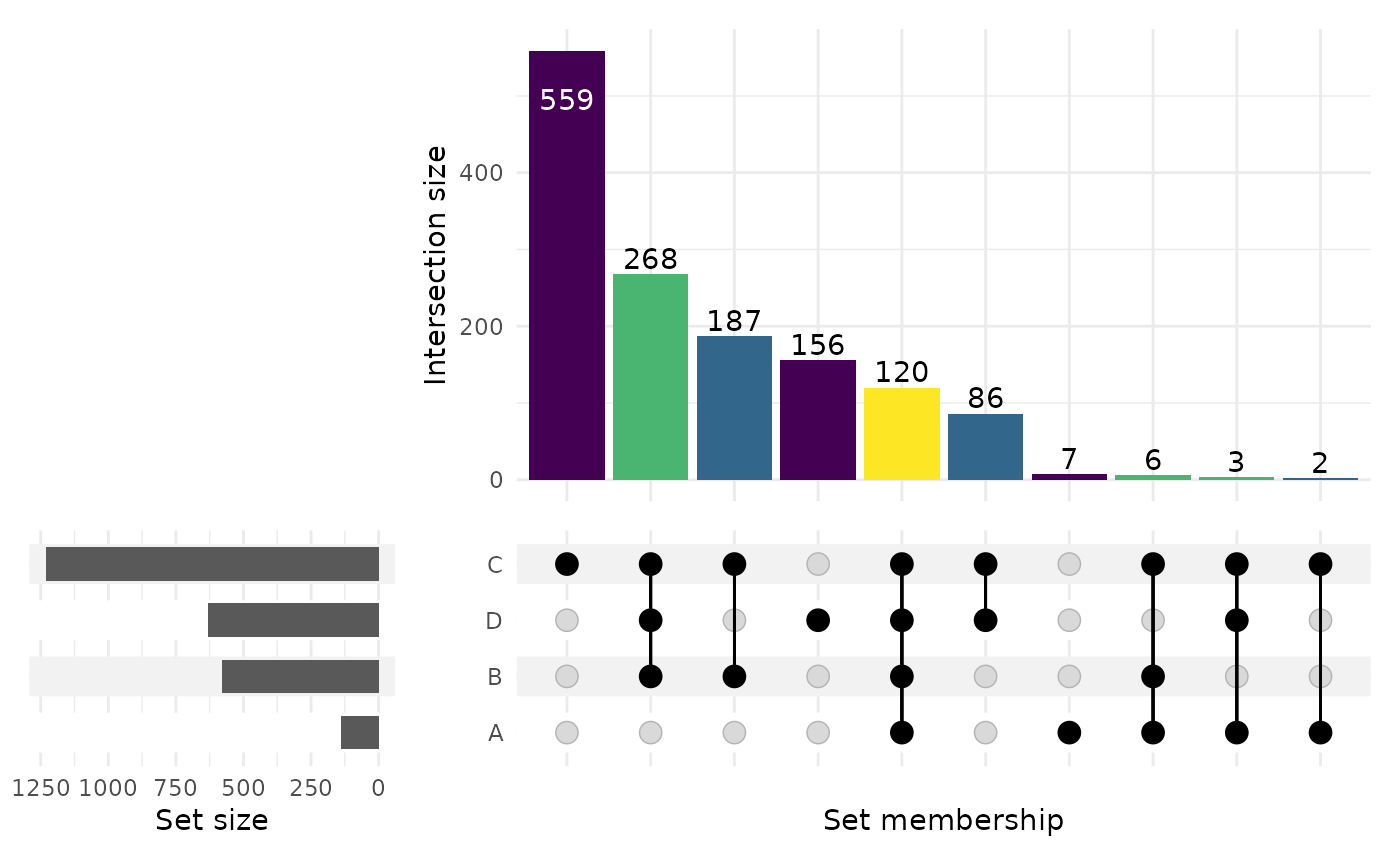
(The chunk above is gated on R >= 4.6 because the
CRAN release of ComplexUpset (1.3.3) is incompatible with
ggplot2 >= 4.0 on older R — see
?vennDiagramLab::render_upset for context.)
Top significant intersections
broom::tidy() returns one row per set pair, with all
five pairwise metrics plus the BH-FDR-adjusted hypergeometric
p-value:
top_pairs <- broom::tidy(result)
top_pairs[order(top_pairs$p_adjusted), c("set_a", "set_b", "intersection",
"jaccard", "p_adjusted",
"significant")]
#> # A tibble: 6 × 6
#> set_a set_b intersection jaccard p_adjusted significant
#> <chr> <chr> <int> <dbl> <dbl> <lgl>
#> 1 COSMIC_CGC OncoKB 581 0.472 0 TRUE
#> 2 COSMIC_CGC IntOGen 388 0.470 0 TRUE
#> 3 OncoKB IntOGen 477 0.344 0 TRUE
#> 4 Vogelstein COSMIC_CGC 126 0.212 1.01e-183 TRUE
#> 5 Vogelstein IntOGen 123 0.190 5.54e-171 TRUE
#> 6 Vogelstein OncoKB 131 0.106 3.13e-151 TRUEEvery pair is significant at FDR < 0.05 (as expected — these catalogs are designed to overlap on biology).
Item-level annotation
broom::augment() returns one row per gene with
set-membership flags and the region label.
gene_table <- broom::augment(result)
head(gene_table)
#> # A tibble: 6 × 6
#> item Vogelstein COSMIC_CGC OncoKB IntOGen region_label
#> <chr> <lgl> <lgl> <lgl> <lgl> <chr>
#> 1 A1CF FALSE FALSE TRUE FALSE C
#> 2 AAMP FALSE FALSE TRUE FALSE C
#> 3 ABCB1 FALSE FALSE TRUE FALSE C
#> 4 ABCC3 FALSE FALSE TRUE FALSE C
#> 5 ABCC4 FALSE FALSE FALSE TRUE D
#> 6 ABI1 FALSE TRUE TRUE FALSE BC
nrow(gene_table) # total unique genes across all four sets
#> [1] 20000
table(gene_table$region_label) # how many genes in each region
#>
#> A ABC ABCD AC ACD BC BCD C CD D
#> 18606 7 6 120 2 3 187 268 559 86 156Save the region summary
to_region_summary_tsv(result, "cancer_drivers_regions.tsv")What’s next
-
vignette("v05_statistics_deep_dive")— interpret the Jaccard / Dice / hypergeometric numbers in detail. -
vignette("v07_pdf_reports")— turn this analysis into a multi-page PDF. -
vignette("v08_custom_styling_and_export")— customize colors, embed in a ggplot, export to PDF/PNG.